Judith L. Campbell

Mechanisms and Regulation of DNA Replication and Repair
A hallmark of cancer cells, in addition to uncontrolled proliferation, is genomic instability, which appears in the form of chromosome loss or gain, gross chromosomal rearrangements, deletions, or amplifications. The mechanisms that suppress such instability are of the utmost interest in understanding the pathogenesis and treatment of cancer. Our lab studies the components of the DNA replication apparatus that promote genomic stability. We use yeast genetics and biochemistry, Xenopus egg extracts, and human cells.

DNA replication is the central process of all actively dividing cells. Blocking this process can result in cell cycle arrest, senescence, and apoptosis. Therefore, DNA replication forks constitute the targets of most cancer chemotherapeutics, including agents that induce DNA lesions, such as camptothecin and cisplatin and ionizing radiation, plus those that stall replication, such as gemcitabine and 5-fluorouracil. If not repaired, this DNA damage may block or collapse DNA replication forks and kill cancer cells. Besides the problem of collateral damage to non-tumor cells, a serious drawback of these therapeutic treatments is that sooner or later the cancer cell may become resistant to the radiation or chemotherapy. Reasons for resistance include increased tolerance for DNA lesions and enhanced capacity for DNA damage response and repair. Therefore, inhibition of proteins that function at the DNA replication/DNA repair interface are attractive targets for sensitizing tumor cells to chemotherapeutic agents. Our intensive studies of DNA2 suggest that it is an Achilles heel for cancer cells, and much of our effort are is aimed at developing small molecule inhibitors to exploit this vulnerability.
At least seven human diseases characterized by cancer predisposition and/or premature aging are correlated with defects in genes encoding DNA helicases. The yeast genome contains 134 open reading frames with helicase motifs, only a few of which have been characterized. Martin Budd in our laboratory identified the first eukaryotic helicase essential for DNA replication, Dna2. He showed by interaction studies that it was a component of the machine that is required for accurate processing of Okazaki fragments during lagging-strand DNA replication. Enzymatic studies to elucidate the sequential action of the DNA polymerases, helicases, and nucleases required for this processing constitute an ongoing mechanistic biochemistry project in the laboratory. Okazaki fragment processing represents the heart of the replication machine, and our studies have revealed that, as in prokaryotes, the replisome is not a machine made up of dedicated parts like its namesake the ribosome. Instead, the replisome is a dynamic structure with proteins constantly exchanging protein and DNA partners to coordinate the rapid and high fidelity synthesis of the anti-parallel leading and lagging strands of the DNA template. Our current work focuses on the regulation, by reversible acetylation and phosphorylation, of the protein/protein and protein/DNA hand-offs that we have defined over the last decade.
One model of cellular aging suggests that accumulation of DNA damage leads to replicative senescence. Most endogenous damage occurs during S phase and leads to replication fork stress. At least three human diseases of premature aging or cancer predisposition - Werner, Bloom, and Rothmund-Thompson - are caused by defects in helicases that interact with Dna2. We found that dna2 mutants have a significantly reduced life span. Microarray analysis showed that the dna2 mutants age by the same pathway as wildtype cells; they just age faster. Interestingly, the human Bloom and Werner genes complement the replication defect of dna2 mutants, suggesting that Dna2 works in the same pathway with these genes. We have now shown that the Dna2 helicase works with the yeast BLM ortholog, Sgs1, in the major pathway of double-strand break repair in yeast and are studying the same process in both yeast and human cells. Together Dna2 and Sgs1 are involved in the initial resection of the 5' terminated strand of the DSB to produce a single-stranded 3' end. This is a crucial step because it is where the cell decides whether to pursue the relatively error-free homologous recombination pathway or the more error-prone non-homologous end-joining repair. The 3' end generated by Dna2/Sgs1 is involved in strand invasion of the homolog and thus, the initiation of strand exchange. Perhaps even more important the single-stranded DNA is a key intermediate in the activation of the cell cycle checkpoint that protects the cell from genome instability in the presence of a double-strand break arising from replication fork failure. In collaboration with Dunphy lab, we readily showed that Dna2 also participates in resection in Xenopus egg extracts. We have now reconstituted the recombination machine both from purified yeast proteins and from purified human counterparts, including Dna2 and BLM helicase. BLM helicase is defective in one of the most cancer-prone diseases yet described, Bloom syndrome. Cells from these patients show a high frequency of sister chromatid exchanges and quadriradials. The biochemical approach provides a mechanistic basis for this dynamic recombination processing machine. Especially for the human proteins, this provides insights previously unavailable due to the difficulty of performing recombination experiments in human cells.
Telomeres, i.e., the ends of linear chromosomes, are a special case of the type of ends found at DSBs. Not surprisingly, Dna2 also plays a significant role at telomeres. In fact, the bulk of Dna2 is localized to telomeres and in yeast, this localization is dynamic. During G1 and G2 phases of the cell cycle, Dna2 is at telomeres. During S phase Dna2 leaves telomeres and is present on the replicating chromatin. Dna2 is also mobilized from telomeres in response to the induction of intrachromosomal double-strand breaks with agents such as bleomycin. At the end of S phase, telomeres become single-stranded in all organisms and this occurs through 5' resection to produce single-stranded 3' overhangs. We have now shown that Dna2 is one of the major enzymes involved in resection at telomeres, as well as internal DSBs. It will be important to investigate if the same holds true in human cells with Dna2 knocked down by shRNA.

{kind=link}
{kind=link}
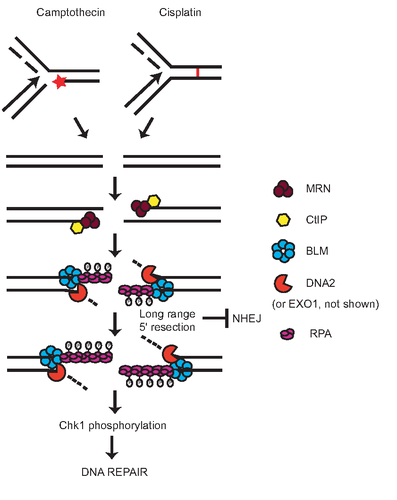
Supplementary Figure 1: Model for DNA end resection after replication stress. Camptothecin or cisplatin exposure blocks replication due to formation of topoisomerase-DNA adducts (red star) or interstand cross links (red link between strands), respectively. Approaching replication forks are unable to proceed past the lesions and may subsequently collapse to generate DSBs. DSBs are first processed by MRN (brown circles)/CtIP (yellow hexagon) to generate short 3' ssDNA. BLM (blue circles), DNA2 (red pacman) or EXO1 (not shown) are necessary for long range resection to produce ssDNA that is capable of binding RPA (purple oblongs). Long range resection is also needed to effect an ATM to ATR switch. RPA bound to DNA is hyperphosphorylated thus promoting ATR phosphorylation of Chk1, induction of cell cycle checkpoint and efficient DNA damage repair. Long range resection precludes the engagement of the NHEJ pathway by preventing the hyperphosphorylation of DNA-PKcs.